Background: White-Sutton Syndrome (WSS) is a rare autosomal dominant neurodevelopmental disorder caused by pathogenic variants in the POGZ gene, which is essential for chromatin remodeling and neuronal development. Because of its broad phenotypic heterogeneity and lack of disease-specific features, early diagnosis and management remain challenging. Timely genetic testing can significantly aid in early diagnosis and intervention, improving patient outcomes. Objective: To describe the clinical and genetic findings of a Chinese pediatric patient with a novel POGZ mutation, summarize the diagnostic approach, and underscore the importance of early genetic testing and multidisciplinary management for the diagnosis and management of WSS. Method: A 4-month-old male infant presented with developmental delay and abnormal liver function. Comprehensive clinical, imaging, auditory, and ophthalmologic evaluations were performed. Whole-genome sequencing and Sanger validation were conducted, followed by multidisciplinary management including nutritional therapy and early rehabilitation. Result: The patient exhibited microcephaly, hypotonia, distinctive facial dysmorphism, auditory impairment, and retinitis pigmentosa. Brain MRI revealed hypoplasia of the corpus callosum. A novel heterozygous frameshift mutation c.2699_2700dup (p.Leu901TyrfsTer2) in POGZ was identified and classified as pathogenic according to ACMG criteria (PVS1 + PS2 + PM2 + PP4). The variant was not reported in existing genetic databases, representing a novel pathogenic mutation expanding the POGZ mutational spectrum. Despite multidisciplinary rehabilitation, neurodevelopmental progress remained limited. Conclusion: This report documents the first Chinese case of White-Sutton Syndrome caused by a novel POGZ frameshift mutation, emphasizing the importance of early genetic testing for accurate diagnosis and timely intervention. Genetic diagnosis combined with personalized rehabilitation may improve long-term neurodevelopmental outcomes for patients with WSS. This case study expands the mutation spectrum of the POGZ gene and provides valuable reference for the diagnosis, treatment, prognosis assessment, and genetic counseling of WSS patients.
| Published in | Clinical Medicine Research (Volume 14, Issue 6) |
| DOI | 10.11648/j.cmr.20251406.13 |
| Page(s) | 223-228 |
| Creative Commons |
This is an Open Access article, distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution and reproduction in any medium or format, provided the original work is properly cited. |
| Copyright |
Copyright © The Author(s), 2025. Published by Science Publishing Group |
White-Sutton Syndrome, POGZ, Novel Mutation, Chromatin Remodeling, Neurodevelopmental Disorder, Case Report
| [1] | Batzir, A. N., White, J., & Sutton, V. R. White-Sutton syndrome [J]. GeneReviews®. Seattle: University of Washington, 2024, (00): 1-3. |
| [2] | Batzir, A. N., Posey, J. E., Song, X., et al. Phenotypic expansion of POGZ-related intellectual disability syndrome (White-Sutton syndrome) [J]. American Journal of Medical Genetics Part A, 2020, 182(1): 38-52. |
| [3] | Murch, O., Jain, V., Benneche, A., et al. Further delineation of the clinical spectrum of White-Sutton syndrome: 12 new individuals and a review of the literature [J]. European Journal of Human Genetics, 2022, 30(1): 95-100. |
| [4] | Nagy, D., Verheyen, S., Wigby, K. M., et al. Genotype-phenotype comparison in POGZ-related neurodevelopmental disorders by using clinical scoring [J]. Genes (Basel), 2022, 13(1): 154. |
| [5] | Bartholomeeusen, K., Christ, F., Hendrix, J., et al. Lens epithelium-derived growth factor/p75 interacts with the transposase-derived DDE domain of POGZ [J]. Journal of Biological Chemistry, 2009, 284(17): 11467–11477. |
| [6] | Dentici, M. L., Niceta, M., Pantaleoni, F., et al. Expanding the phenotypic spectrum of truncating POGZ mutations: Association with CNS malformations, skeletal abnormalities, and distinctive facial dysmorphism [J]. American Journal of Medical Genetics Part A, 2017, 173(7): 1965–1969. |
| [7] | White, J., Beck, C. R., Harel, T., et al. POGZ truncating alleles cause syndromic intellectual disability [J]. Genome Medicine, 2016, 8(1): 3. |
| [8] | Wang, T., Guo, H., Xiong, B., et al. De novo genic mutations among a Chinese autism spectrum disorder cohort [J]. Nature Communications, 2016, 7: 13316. |
| [9] | Heath, J., Cheyou, E. S., Findlay, S., et al. POGZ promotes homology-directed DNA repair in an HP1-dependent manner [J]. EMBO Reports, 2022, 23(1): e51041. |
| [10] | Metcalf, C., Waller, D. P., Nie, Y., et al. Genotype–Phenotype Comparison in POGZ-Related Neurodevelopmental Disorders. Genes. 2022, 13(1), 154. |
| [11] | White, S. M., Todd, E. J., Mahmood, S., et al. Further Delineation of the Clinical Spectrum of White–Sutton Syndrome: 12 New Individuals and a Review of the Literature. European Journal of Human Genetics. 2021, 29(10), 1517–1527. |
| [12] | Hamada, N., Nishijo, T., Iwamoto, I., et al. Analyses of conditional knockout mice for POGZ, a gene responsible for neurodevelopmental disorders in excitatory and inhibitory neurons in the brain [J]. Cells, 2024, 13(6): 540. |
| [13] | Deng, L., Mojica-Perez, S. P., Azaria, R. D., et al. Loss of POGZ alters neural differentiation of human embryonic stem cells [J]. Molecular and Cellular Neuroscience, 2022, 120: 103727. |
| [14] | Mudassir, B. U., Mudassir, M., Williams, J. B., & Agha, Z. Denovo variants in POGZ and YY1 genes: The novel mega players for neurodevelopmental syndromes in two unrelated consanguineous families [J]. PLoS One, 2025, 20(1): e0315597. Published 2025 Jan 8. |
| [15] | Leibovitch, L., Gorenshtein, A., Bibi, E., & Uri, A. Malignant catatonia in an adolescent with pogo transposable element derived with zinc finger domain (POGZ) gene mutation: case report [J]. BJPsych Open, 2025, 11(5): e170. Published 2025 Aug 1. |
| [16] | Ferretti, A., Barresi, S., Trivisano, M., et al. POGZ-related epilepsy: Case report and review of the literature [J]. American Journal of Medical Genetics Part A, 2019, 179(8): 1631-1636. |
| [17] | Giraldo-Ocampo, S., Pacheco-Orozco, R. A., Pachajoa, H. A Novel POGZ Variant in a Patient with Intellectual Disability and Obesity [J]. Applied Clinical Genetics. 2022, 15: 63-68. Published 2022 Jul 6. |
| [18] | Caffarelli, C., Santamaria, F., Piro, E., et al. New insights in pediatrics in 2021: choices in allergy and immunology, critical care, endocrinology, gastroenterology, genetics, haematology, infectious diseases, neonatology, neurology, nutrition, palliative care, respiratory tract illnesses and telemedicine [J]. Italian Journal of Pediatrics, 2022, 48(1): 189. Published 2022 Nov 26. |
| [19] | Bar, O., Ebenau, L., Weiner, K., Mintz, M., & Boles, R. G. Whole exome/genome sequencing in cyclic vomiting syndrome reveals multiple candidate genes, suggesting a model of elevated intracellular cations and mitochondrial dysfunction [J]. Frontiers in Neurology. 2023 May 5; 14: 1151835. |
APA Style
Mo, Z., Zeng, J., Pan, X., Song, Y. (2025). Clinical and Genetic Analysis of a Chinese Patient Carrying a Novel POGZ Variant Associated with White-Sutton Syndrome. Clinical Medicine Research, 14(6), 223-228. https://doi.org/10.11648/j.cmr.20251406.13
ACS Style
Mo, Z.; Zeng, J.; Pan, X.; Song, Y. Clinical and Genetic Analysis of a Chinese Patient Carrying a Novel POGZ Variant Associated with White-Sutton Syndrome. Clin. Med. Res. 2025, 14(6), 223-228. doi: 10.11648/j.cmr.20251406.13
AMA Style
Mo Z, Zeng J, Pan X, Song Y. Clinical and Genetic Analysis of a Chinese Patient Carrying a Novel POGZ Variant Associated with White-Sutton Syndrome. Clin Med Res. 2025;14(6):223-228. doi: 10.11648/j.cmr.20251406.13
@article{10.11648/j.cmr.20251406.13,
author = {Zhi-Jun Mo and Jiang-Mei Zeng and Xiang Pan and Yuan-Zong Song},
title = {Clinical and Genetic Analysis of a Chinese Patient Carrying a Novel POGZ Variant Associated with White-Sutton Syndrome},
journal = {Clinical Medicine Research},
volume = {14},
number = {6},
pages = {223-228},
doi = {10.11648/j.cmr.20251406.13},
url = {https://doi.org/10.11648/j.cmr.20251406.13},
eprint = {https://article.sciencepublishinggroup.com/pdf/10.11648.j.cmr.20251406.13},
abstract = {Background: White-Sutton Syndrome (WSS) is a rare autosomal dominant neurodevelopmental disorder caused by pathogenic variants in the POGZ gene, which is essential for chromatin remodeling and neuronal development. Because of its broad phenotypic heterogeneity and lack of disease-specific features, early diagnosis and management remain challenging. Timely genetic testing can significantly aid in early diagnosis and intervention, improving patient outcomes. Objective: To describe the clinical and genetic findings of a Chinese pediatric patient with a novel POGZ mutation, summarize the diagnostic approach, and underscore the importance of early genetic testing and multidisciplinary management for the diagnosis and management of WSS. Method: A 4-month-old male infant presented with developmental delay and abnormal liver function. Comprehensive clinical, imaging, auditory, and ophthalmologic evaluations were performed. Whole-genome sequencing and Sanger validation were conducted, followed by multidisciplinary management including nutritional therapy and early rehabilitation. Result: The patient exhibited microcephaly, hypotonia, distinctive facial dysmorphism, auditory impairment, and retinitis pigmentosa. Brain MRI revealed hypoplasia of the corpus callosum. A novel heterozygous frameshift mutation c.2699_2700dup (p.Leu901TyrfsTer2) in POGZ was identified and classified as pathogenic according to ACMG criteria (PVS1 + PS2 + PM2 + PP4). The variant was not reported in existing genetic databases, representing a novel pathogenic mutation expanding the POGZ mutational spectrum. Despite multidisciplinary rehabilitation, neurodevelopmental progress remained limited. Conclusion: This report documents the first Chinese case of White-Sutton Syndrome caused by a novel POGZ frameshift mutation, emphasizing the importance of early genetic testing for accurate diagnosis and timely intervention. Genetic diagnosis combined with personalized rehabilitation may improve long-term neurodevelopmental outcomes for patients with WSS. This case study expands the mutation spectrum of the POGZ gene and provides valuable reference for the diagnosis, treatment, prognosis assessment, and genetic counseling of WSS patients.},
year = {2025}
}
TY - JOUR T1 - Clinical and Genetic Analysis of a Chinese Patient Carrying a Novel POGZ Variant Associated with White-Sutton Syndrome AU - Zhi-Jun Mo AU - Jiang-Mei Zeng AU - Xiang Pan AU - Yuan-Zong Song Y1 - 2025/12/17 PY - 2025 N1 - https://doi.org/10.11648/j.cmr.20251406.13 DO - 10.11648/j.cmr.20251406.13 T2 - Clinical Medicine Research JF - Clinical Medicine Research JO - Clinical Medicine Research SP - 223 EP - 228 PB - Science Publishing Group SN - 2326-9057 UR - https://doi.org/10.11648/j.cmr.20251406.13 AB - Background: White-Sutton Syndrome (WSS) is a rare autosomal dominant neurodevelopmental disorder caused by pathogenic variants in the POGZ gene, which is essential for chromatin remodeling and neuronal development. Because of its broad phenotypic heterogeneity and lack of disease-specific features, early diagnosis and management remain challenging. Timely genetic testing can significantly aid in early diagnosis and intervention, improving patient outcomes. Objective: To describe the clinical and genetic findings of a Chinese pediatric patient with a novel POGZ mutation, summarize the diagnostic approach, and underscore the importance of early genetic testing and multidisciplinary management for the diagnosis and management of WSS. Method: A 4-month-old male infant presented with developmental delay and abnormal liver function. Comprehensive clinical, imaging, auditory, and ophthalmologic evaluations were performed. Whole-genome sequencing and Sanger validation were conducted, followed by multidisciplinary management including nutritional therapy and early rehabilitation. Result: The patient exhibited microcephaly, hypotonia, distinctive facial dysmorphism, auditory impairment, and retinitis pigmentosa. Brain MRI revealed hypoplasia of the corpus callosum. A novel heterozygous frameshift mutation c.2699_2700dup (p.Leu901TyrfsTer2) in POGZ was identified and classified as pathogenic according to ACMG criteria (PVS1 + PS2 + PM2 + PP4). The variant was not reported in existing genetic databases, representing a novel pathogenic mutation expanding the POGZ mutational spectrum. Despite multidisciplinary rehabilitation, neurodevelopmental progress remained limited. Conclusion: This report documents the first Chinese case of White-Sutton Syndrome caused by a novel POGZ frameshift mutation, emphasizing the importance of early genetic testing for accurate diagnosis and timely intervention. Genetic diagnosis combined with personalized rehabilitation may improve long-term neurodevelopmental outcomes for patients with WSS. This case study expands the mutation spectrum of the POGZ gene and provides valuable reference for the diagnosis, treatment, prognosis assessment, and genetic counseling of WSS patients. VL - 14 IS - 6 ER -
Department of Pediatrics, The First Affiliated Hospital of Jinan University, Guangzhou, China
Department of Pediatrics, Dongguan People's Hospital, Dongguan, China
Department of Pediatrics, The First Affiliated Hospital of Jinan University, Guangzhou, China
Department of Pediatrics, The First Affiliated Hospital of Jinan University, Guangzhou, China
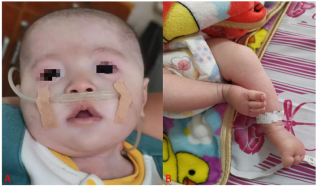
Figure 1. Craniofacial deformity characteristics.
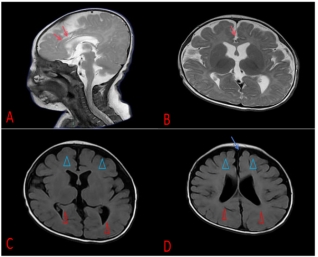
Figure 2. Brain MRI of the child. The corpus callosum at the genu and body (indicated by red arrows), bilateral frontal lobes (indicated by blue triangles), and temporal lobes show underdevelopment (indicated by red triangles). The corresponding extra-axial spaces are slightly widened (indicated by blue arrows), suggesting possible cerebral dysgenesis.
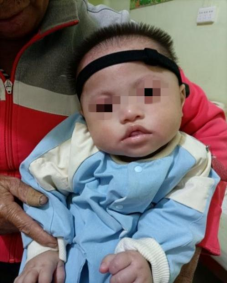
Figure 3. Follow-up at 1 year and 1 month of age. The child is wearing a hearing aid, has weak neck control, cannot sit, responds to calling with vocalizations, and smiles.
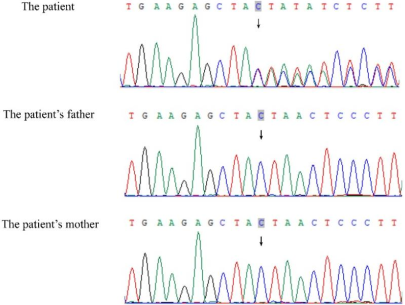
Figure 4. Sanger sequencing of the POGZ gene in a family of three. The child exhibits a heterozygous mutation in the POGZ gene, c.2699_2700dup (p.Leu901TyrfsTer2), which is not present in either of the parents.

