Gaucher disease (GD) is an autosomal recessive lysosomal storage disorder caused by variants in the GBA gene. This study reports a novel pathogenic large deletion in the GBA gene identified in a 70-day-old male infant presenting with cholestasis and hepatosplenomegaly, leading to a diagnosis of GD. Comprehensive genetic analysis using whole-exome sequencing (WES) revealed compound heterozygous variants, a maternally inherited c.1448T>C (p.Leu483Pro) missense variant and a paternally derived large deletion encompassing both the GBAP1 pseudogene and the functional GBA gene. The paternal origin of the deletion was confirmed by quantitative PCR (qPCR), and long-range PCR with subsequent sequencing precisely mapped the breakpoints, characterizing the deletion as 20,627 bp in length. A critical diagnostic finding was that standard Sanger sequencing initially failed to detect this deletion, misleadingly suggesting the infant was homozygous for the missense variant. This case highlights a significant limitation of Sanger sequencing, which can misinterpret large heterozygous deletions as false homozygosity due to allele dropout. Consequently, this report underscores the necessity of employing comprehensive genomic methods like WES as a first-line diagnostic test for lysosomal storage disorders such as GD, ensuring accurate detection of complex variants including large structural variants.
| Published in | Clinical Medicine Research (Volume 14, Issue 6) |
| DOI | 10.11648/j.cmr.20251406.15 |
| Page(s) | 238-243 |
| Creative Commons |
This is an Open Access article, distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution and reproduction in any medium or format, provided the original work is properly cited. |
| Copyright |
Copyright © The Author(s), 2025. Published by Science Publishing Group |
Gaucher Disease, Gaucher I, GBA Gene, Paternal Large Deletion, Whole-exome Sequencing, Enzyme Replacement Therapy
Indices (range) | Ages | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
49D | 52D | 66D | 67D | 70D | 72D | 76D | 81D | 84D | 284D | |
ALT (5-40U/L) | 54.6 | 66 | 192.3 | 302 | 256 | 220 | 618 | 449 | 359 | 60 |
AST (5-40U/L) | 117.5 | 122 | 278.1 | 331 | 311 | 285 | 776 | 448 | 289 | 64 |
GGT (8-50U/L) | 189.6 | 122 | 108.9 | 103 | 92 | 74 | 80 | 74 | 70 | 15 |
TBIL (2-19μmol/L) | 59.9 | 57.5 | 31.2 | 36.6 | 30.4 | 22.9 | 23.5 | 17.5 | 15.3 | 4.9 |
DBIL (0-6μmol/L) | 48.6 | 35.4 | 22.9 | 22.8 | 23.2 | 16 | 16.5 | 12.3 | 9.8 | 1.7 |
IBIL (2.56-20.9μmol/L) | 11.3 | 22.1 | 8.3 | 13.8 | 7.2 | 6.9 | 7 | 5.2 | 5.5 | 3.2 |
TBA (0-10μmol/L) | 57.56 | 69.2 | 104.54 | 113.2 | 96.6 | 44.7 | 42.9 | 74.9 | 27.1 | 3.8 |
GD | Gaucher Disease |
WES | Whole-exome Sequencing |
GCase | Glucocerebrosidase |
GlcCer | Glucosylceramide |
HGMD | Human Gene Vatiant Database |
ERT | Enzyme Replacement Therapy |
qPCR | Quantitative Real-time Polymerase Chain Reaction |
ACMG | American College of Medical Genetics and Genomics |
NGS | Next-generation Sequencing |
NAHRCNV | Non-allelic Homologous Recombination Copy Number Variation |
| [1] | Stirnemann J, Belmatoug N, Camou F, Serratrice C, Froissart R, Caillaud C, et al. A Review of Gaucher Disease Pathophysiology, Clinical Presentation and Treatments. International Journal of Molecular Sciences. 2017, 18(2), 441. |
| [2] | Beutler E. Gaucher Disease. Advances in Genetics. 1995, 32, 17–49. |
| [3] | Ankleshwaria C, Mistri M, Bavdekar A, Tamhankar P, Sheth J, Sanghavi D, et al. Novel Variants in the Glucocerebrosidase Gene of Indian Patients with Gaucher Disease. Journal of Human Genetics. 2014, 59(4), 223–228. |
| [4] | Charrow J, Andersson HC, Kaplan P, Kolodny EH, Mistry P, Pastores G, et al. The Gaucher Registry: Demographics and Disease Characteristics of 1698 Patients with Gaucher Disease. Archives of Internal Medicine. 2000, 160(18), 2835–2843. |
| [5] | Zhang TB, Wen XL, Zhang XL, Wang JC, Wang XH, Zhang AM. Genetic and Clinical Analysis of 20 Cases with Gaucher Disease. Chinese Journal of Hematology. 2024, 45(1), 82–85. |
| [6] | Dimitriou E, Moraitou M, Cozar M, Dardioti M, Mintzaki I, Giannakoulas G, et al. Gaucher Disease: Biochemical and Molecular Findings in 141 Patients Diagnosed in Greece. Molecular Genetics and Metabolism Reports. 2020, 24, 100614. |
| [7] | Choy FY, Zhang W, Shi HP, Zay A, Campbell T, Tang N, et al. Gaucher Disease among Chinese Patients: Review on Genotype/Phenotype Correlation from 29 Patients and Identification of Novel and Rare Alleles. Blood Cells, Molecules, and Diseases. 2007, 38(3), 287–293. |
| [8] | Feng Y, Huang Y, Tang C, Wu Y, Chen X, Zhang L, et al. Clinical and Molecular Characteristics of Patients with Gaucher Disease in Southern China. Blood Cells, Molecules, and Diseases. 2018, 68, 30–34. |
| [9] | Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine. 2015, 17(5), 405–424. |
| [10] | Gu W, Zhang F, Lupski JR. Mechanisms for Human Genomic Rearrangements. Pathogenetics. 2008, 1(1), 4. |
| [11] | Horowitz M, Pasmanik-Chor M, Ron I, Kolodny EH. The Enigma of the E326K Variant in Acid β-Glucocerebrosidase. Molecular Genetics and Metabolism. 2011, 104(1–2), 35–38. |
| [12] | Poll LW, Cox ML, Godehardt E, Steinhof V, vom Dahl S. Whole Body MRI in Type I Gaucher Patients: Evaluation of Skeletal Involvement. Blood Cells, Molecules, and Diseases. 2011, 46(1), 53–59. |
| [13] | Costagliola G, De Marco E, Massei F, Di Fusco C, Notarangelo LD, Moriondo M, et al. The Etiologic Landscape of Lymphoproliferation in Childhood: Proposal for a Diagnostic Approach Exploring from Infections to Inborn Errors of Immunity and Metabolic Diseases. Therapeutics and Clinical Risk Management. 2024, 20, 261–274. |
| [14] | Barbier C, Devisme L, Dobbelaere D, Noizet O, Nelken B, Gottrand F. Neonatal Cholestasis and Infantile Gaucher Disease: A Case Report. Acta Paediatrica. 2002, 91(12), 1399–1401. |
| [15] | Hirachan R, Horman A, Burke D, Heales S. Evaluation, in a Highly Specialised Enzyme Laboratory, of a Digital Microfluidics Platform for Rapid Assessment of Lysosomal Enzyme Activity in Dried Blood Spots. JIMD Reports. 2024, 65(2), 124–131. |
| [16] | Marshall CR, Chowdhury S, Taft RJ, Lebo MS, Buchan JG, Harrison SM, et al. Best Practices for the Analytical Validation of Clinical Whole-Genome Sequencing Intended for the Diagnosis of Germline Disease. NPJ Genomic Medicine. 2020, 5, 47. |
| [17] | Kosicki M, Tomberg K, Bradley A. Repair of Double-Strand Breaks Induced by CRISPR–Cas9 Leads to Large Deletions and Complex Rearrangements. Nature Biotechnology. 2018, 36(8), 765–771. |
| [18] | Adams DR, Eng CM. Next-Generation Sequencing to Diagnose Suspected Genetic Disorders. New England Journal of Medicine. 2018, 379(14), 1353–1362. |
| [19] | Zampieri, S., Cattarossi, S., Pavan, E., Barbato, A., Fiumara, A., Peruzzo, P., Scarpa, M., Ciana, G., Dardis, A. Accurate Molecular Diagnosis of Gaucher Disease Using Clinical Exome Sequencing as a First-Tier Test. International Journal of Molecular Sciences. 2021, 22(11), 5538. |
| [20] | Heinz N, Vittorio J. Treatment of Cholestasis in Infants and Young Children. Current Gastroenterology Reports. 2023, 25(11), 344–354. |
| [21] | Komlosi K, Solyom A, Beck M. The Role of Next-Generation Sequencing in the Diagnosis of Lysosomal Storage Disorders. Journal of Inborn Errors of Metabolism and Screening. 2016, 4, 232640981666937. |
APA Style
Shen, K., Liao, P., Song, Y. (2025). Identification of a Novel GBA Deletion Spanning 20,627bp Associated with Gaucher Disease: A Case Report. Clinical Medicine Research, 14(6), 238-243. https://doi.org/10.11648/j.cmr.20251406.15
ACS Style
Shen, K.; Liao, P.; Song, Y. Identification of a Novel GBA Deletion Spanning 20,627bp Associated with Gaucher Disease: A Case Report. Clin. Med. Res. 2025, 14(6), 238-243. doi: 10.11648/j.cmr.20251406.15
AMA Style
Shen K, Liao P, Song Y. Identification of a Novel GBA Deletion Spanning 20,627bp Associated with Gaucher Disease: A Case Report. Clin Med Res. 2025;14(6):238-243. doi: 10.11648/j.cmr.20251406.15
@article{10.11648/j.cmr.20251406.15,
author = {Ke-yuan Shen and Phoebe Liao and Yuan-Zong Song},
title = {Identification of a Novel GBA Deletion Spanning 20,627bp Associated with Gaucher Disease: A Case Report},
journal = {Clinical Medicine Research},
volume = {14},
number = {6},
pages = {238-243},
doi = {10.11648/j.cmr.20251406.15},
url = {https://doi.org/10.11648/j.cmr.20251406.15},
eprint = {https://article.sciencepublishinggroup.com/pdf/10.11648.j.cmr.20251406.15},
abstract = {Gaucher disease (GD) is an autosomal recessive lysosomal storage disorder caused by variants in the GBA gene. This study reports a novel pathogenic large deletion in the GBA gene identified in a 70-day-old male infant presenting with cholestasis and hepatosplenomegaly, leading to a diagnosis of GD. Comprehensive genetic analysis using whole-exome sequencing (WES) revealed compound heterozygous variants, a maternally inherited c.1448T>C (p.Leu483Pro) missense variant and a paternally derived large deletion encompassing both the GBAP1 pseudogene and the functional GBA gene. The paternal origin of the deletion was confirmed by quantitative PCR (qPCR), and long-range PCR with subsequent sequencing precisely mapped the breakpoints, characterizing the deletion as 20,627 bp in length. A critical diagnostic finding was that standard Sanger sequencing initially failed to detect this deletion, misleadingly suggesting the infant was homozygous for the missense variant. This case highlights a significant limitation of Sanger sequencing, which can misinterpret large heterozygous deletions as false homozygosity due to allele dropout. Consequently, this report underscores the necessity of employing comprehensive genomic methods like WES as a first-line diagnostic test for lysosomal storage disorders such as GD, ensuring accurate detection of complex variants including large structural variants.},
year = {2025}
}
TY - JOUR T1 - Identification of a Novel GBA Deletion Spanning 20,627bp Associated with Gaucher Disease: A Case Report AU - Ke-yuan Shen AU - Phoebe Liao AU - Yuan-Zong Song Y1 - 2025/12/31 PY - 2025 N1 - https://doi.org/10.11648/j.cmr.20251406.15 DO - 10.11648/j.cmr.20251406.15 T2 - Clinical Medicine Research JF - Clinical Medicine Research JO - Clinical Medicine Research SP - 238 EP - 243 PB - Science Publishing Group SN - 2326-9057 UR - https://doi.org/10.11648/j.cmr.20251406.15 AB - Gaucher disease (GD) is an autosomal recessive lysosomal storage disorder caused by variants in the GBA gene. This study reports a novel pathogenic large deletion in the GBA gene identified in a 70-day-old male infant presenting with cholestasis and hepatosplenomegaly, leading to a diagnosis of GD. Comprehensive genetic analysis using whole-exome sequencing (WES) revealed compound heterozygous variants, a maternally inherited c.1448T>C (p.Leu483Pro) missense variant and a paternally derived large deletion encompassing both the GBAP1 pseudogene and the functional GBA gene. The paternal origin of the deletion was confirmed by quantitative PCR (qPCR), and long-range PCR with subsequent sequencing precisely mapped the breakpoints, characterizing the deletion as 20,627 bp in length. A critical diagnostic finding was that standard Sanger sequencing initially failed to detect this deletion, misleadingly suggesting the infant was homozygous for the missense variant. This case highlights a significant limitation of Sanger sequencing, which can misinterpret large heterozygous deletions as false homozygosity due to allele dropout. Consequently, this report underscores the necessity of employing comprehensive genomic methods like WES as a first-line diagnostic test for lysosomal storage disorders such as GD, ensuring accurate detection of complex variants including large structural variants. VL - 14 IS - 6 ER -
Department of Pediatrics, The First Affiliated Hospital of Jinan University, Guangzhou, China
Department of Pediatrics, The First Affiliated Hospital of Jinan University, Guangzhou, China
Department of Pediatrics, The First Affiliated Hospital of Jinan University, Guangzhou, China
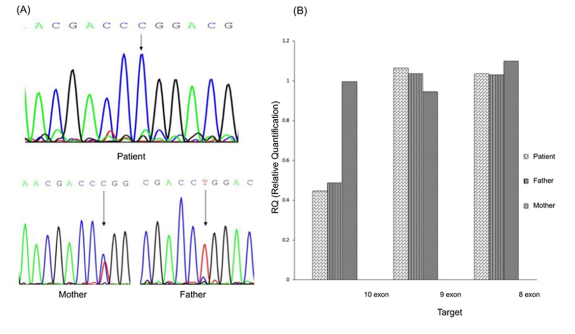
Figure 1. Genetic findings in the GD patient and parents: A hemizygous c.1448T>C (p.Leu483Pro) variant in exon 10 of the GBA gene was detected in the patient. This variant was heterozygous in the mother and wild-type in the father, as confirmed by Sanger sequencing (Figure A). Further real-time PCR analysis of the GBA gene in the family trio showed a normal copy number of exons 8 and 9, but a reduced signal in exon 10 in both the patient and the father, indicating a deletion covering this exon (Figure B). The c.1448T>C (p.Leu483Pro) variant is located within exon 10 of the GBA gene.
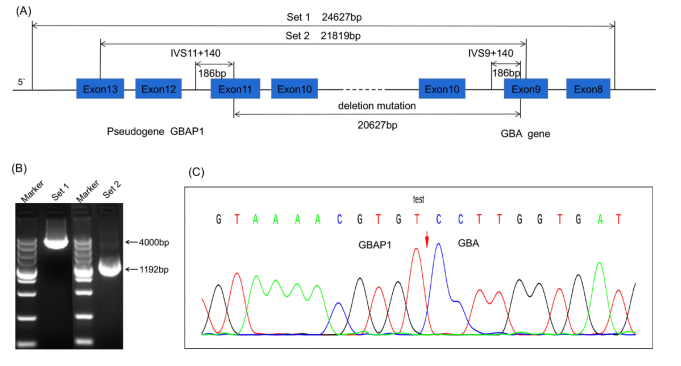
Figure 2. Characterization of a novel large segment deletion involving the GBAP1 and GBA genes. (A) Schematic of the long-range PCR strategy for breakpoint mapping. Two primer sets flanking the predicted deletion region were employed. (B) Electrophoresis of long-range PCR products. The proband and father showed aberrantly shorter fragments (Set 1: ~4,000 bp vs. the expected ~24,627 bp wild-type; Set 2: ~1,192 bp vs. ~21,819 bp), confirming the heterozygous deletion. (C) Sanger sequencing chromatogram of the breakpoint junction from the purified PCR product. Sequence alignment indicated by the vertical dashed line (GRCh37/hg19: chr1: 155184891_155205517), defining a 20,627 bp deletion that includes GBAP1 and exon 10 of GBA.

